MEN syndrome je familiární onemocnění s autozomálně dominantním typem dědičnosti, pro které jsou typické změny v endokrinní funkci s tvorbou řady endokrinních tumorů. Většinou jde o benigní nádory s maligním potenciálem.
MEN syndromy dělíme na MEN typu 1, 2A a 2B.
MEN syndromy dělíme na MEN typu 1, 2A a 2B.
Syndrom MEN 1
(Wermerův syndrom) charakterizuje vznik nádorů příštítných tělísek, předního laloku hypofýzy a buněk Langerhansových ostrůvků. Kromě těchto tumorů se dále často vyskytují duodenální gastrinomy, karcinoidy, nádory nadledvin, štítné žlázy a lipomy.
- MEN1 má autozomálně dominantní charakter dědičnosti.
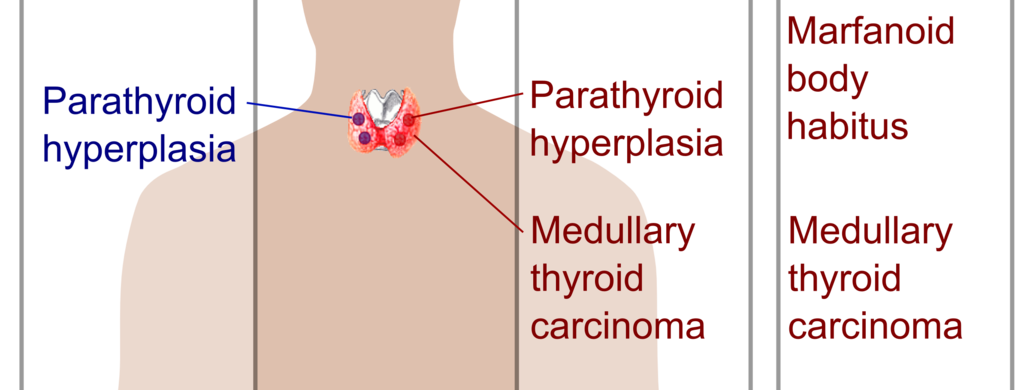
- MEN 2A (Sippleův syndrom) zahrnuje medulární karcinom štítné žlázy, feochromocytom a primární hyperparatyreozu.
- MEN 2B syndrom je charakterizován současným výskytem medulárního karcinomu štítné žlázy a feochromocytomu, nálezem marfanoidního habitu s mukozní ganglioneuromatozou.
- V poslední době byl nově definován syndromMEN 4 s výskytem familiárního izolovaného adenomu hypofýzy. Jde o zcela vzácnýnález, u kterého byla prokázána mutace inhibitoru cyklindependentní kinázy genu p27, lokalizovaném na chromosomu 12p13.
MEN 1
U MEN 1 syndromu bývají postiženy hlavně buňky adenohypofýzy, příštítných tělísek a slinivky břišní, ale byl nalezen i kombinovaný výskyt s dalšími 20 endokrinními i neendokrinními nádory. MEN 1 syndrom je způsobem mutací v tumor supresorovém genu na chromosomu 11q13, kodujícím menin, který je lokalizován v jádře buněk. Tato dědičná, autozomálně dominanní mutace je přítomná ve všech buňkách v těle. Funkce meninu není zatím jednoznačně objasněna, nejspíše se podílí na negativní regulaci buněčné proliferace. Mutace MEN1 genu jsou detekovány u 80–90 % probandů s familiárním syndromem MEN 1. Hyperparatyróza je nejčastějším nálezem, vyskytuje se až v 95 % všech MEN1 syndromů. Asi ve čtvrtině případů MEN1 se setkáme s tumory hypofýzy (prolaktinomy) a ve 40 % případů s enteropankreatickými tumory (především gastrinomem).
Diagnostika
Syndrom MEN1 je charakterizován přítomností nejméně dvou typů výše zmíněných endokrinních tumorů. Hyperparatyreóza u adenomu příštítných tělísek je obvykle prvním klinickým nálezem u MEN1, nejčastěji se vyskytuje u mladých pacientů ve věku 20–25 let. Diagnóza je založena na biochemickém vyšetření (zvýšené hladiny
vápníku a parathormonu) a na lokalizační diagnostice. Hyperkalcémie přispívá rovněž k vzestupu sekrece gastrinu z gastrinomu, který často doprovází adenomy příštítných tělísek a vede k Zollinger-Ellisonovu syndromu (hypersekreci žaludečních kyselin a výskytu duodenálního vředu). U všech pacientů s podezřením na MEN 1 syndrom provádíme jako vstupní zobrazovací vyšetření ultrasonografi i a při podezření na adenom příštítného tělíska je vhodné provedení scintigrafi e 99m
Tc –MIBI. U nádorů hypofýzy volíme jako základní zobrazovací metodu MRI , CT vyšetření břicha pak pro průkaz pankreatických tumorů.
MEN 1 syndrom se také vyskytuje s adrenokortikální hyperplazií (až ve 33–40 %), s karcinoidem, karcinomem štítné žlázy, thymomem bukálními mukozními tumory, polypózou tlustého střeva, hypertrofickou gastritidou, hypoproteinémií a hypochlorhydrémií. Genová analýza je nezbytná pro potvrzení klinického podezření a je nezastupitelná zejména u asymptomatických nemocných a jejich pokrevných příbuzných. Genetický screening by měl být proveden ještě v dětském věku probanda. Pro potvrzení adenomu příštítného tělíska je optimální věk pro screening kolem 8 let, pro průkaz gastrinomu či karcinoidu je vhodné tento screening provést
kolem 20. roku věku. Familiární forma MEN 1 je defi nována jako přítomnost nejméně jednoho pacienta s MEN 1 a nejméně jednoho prvostupňového příbuzného alespoň s jedním ze zmíněných endokrinních nádorů (příštítných tělísek, hypofýzy či gastroenteropankreatického systému) v rodině. Při průkazu zárodečné mutace
MEN1 postačuje jednoorgánové postižení. Pokud byla nalezena u pacienta zárodečná mutace v genu MEN1, doporučuje se provést cílené molekulárně genetické vyšetření u příbuzných rizikových osob. Mutace bývá zjištěna v 60–90 % typických případů. Negativní (neinformativní) výsledek syndrom MEN1 nevylučuje. V ČR genetické vyšetření MEN1 syndromu není zatím Mnohočetná endokrinní neoplázie
(MEN syndrom).
Klinický obraz
Nejčastějším projevem onemocnění je primární hyperparatyreóza, která na rozdíl od sporadické formy nastupuje v ranějším věku, vyskytuje se u obou pohlaví, adenomy jsou mnohočetné a postihují několik příštítných tělísek. Tumory gastroenteropankreatického systému postihují především pankreas a stěnu duodena, bývají rovněž mnohočetné. Imunocytochemicky se prokazuje obvyklevíce hormonů: gastrin, glukagon, inzulin, pankreatický polypeptid. Nejčastěji se klinicky manifestuje gastrinom, se známkami žaludeční hypersekrece s peptickými komplikacemi a průjmy, a inzulinom, u kterého jsou charakteristické hypoglykemie nalačno. Nádory vyskytující se v rámci MEN1 syndromu mívají asi o 10 let časnější nástup než je tomu u sporadických forem. Často metastazují do regionálních lymfatických uzlin, do jater a kostí. Karcinoid, v rámci MEN 1 syndromu, postihuje častěji thymus, bronchus nebo žaludek. Klinicky se projevuje asi v 30–50 % karcinoidovým syndromem – imperativními průjmy s tenesmy a záchvaty zarudnutí kůže, někdy spojené s pocity horka, s nálezem zvýšeného vylučování
kyseliny hydroxyindoloctové, vzácně produkcí ACTH, kalcitoninu, nebo GHRH.
Terapie a prevence
Léčba spočívá v chirurgickém odstranění nádorů a farmakoterapii hormonální ´ nadprodukce. U primární hyperparatyreózy je nezbytné provedení subtotální či totální paratyreoidektomie. U těchto nemocných jsou však časté recidivy do 10 let, proto jsou k operaci indikováni nemocní s výraznou hyperkalcémií a/nebo s kostními či renálními projevy. Gastrinom nebývá, vzhledem k mnohočetnému postižení, obvykle chirurgicky řešitelný a léčba spočívá v podání inhibitorů protonové pumpy. Inzulinomy bývají většinou solitární, proto je zde preferováno chirurgické odstranění většiny pankreatu. U inoperabilních forem se podávají analoga somatostatinu, event. aloxan aj. Léčba nádorů hypofýzy se neliší od léčby sporadických forem.
MEN 2
Jde o vzácné autozomálně dominantní onemocnění, při kterém se vyskytuje medulární karcinom štítné žlázy (MTC) vycházející z parafolikulárních buněk, často v kombinaci s feochromocytomem a hyperplazií či adenomem příštítných tělísek. MEN2 zahrnuje tři klinické varianty onemocnění:
MEN2A – s predispozicí k medulárnímu karcinomu štítné žlázy, primární hyperparatyreóze a feochromocytomu (nejčastější varianta– 95 % případů MEN2)
MEN2B – kde, kromě přítomnosti MTC a feochromocytomu, je typickým nálezem výskyt mnohočetných slizničních neurinomůa marfanoidní habitus.
FMTC – s predispozicí pouze k medulárnímu karcinomu štítné žlázy.
Klinika
První projevem MTC může být uzel ve štítné žláze s typickou lokalizací ve středních partiích žlázy, popř. při jejích horních pólech, kde je fyziologicky nejvyšší koncentrace C-buněk. U nemocných s již hmatným nádorem je časté i metastatické postižení regionálních lymfatických uzlin (u sporadických forem až v 65–70 % případech). Funkce štítné žlázy bývá obvykle normální, struma ani uzliny nebývají palpačně citlivé a přes vysoké hladiny kalcitoninu nebývá zaznamenán pokles hladiny vápníku v krevním séru. Sonograficky se uzel zobrazuje jako hypoechogenní ložisko s kalcifi kacemi, v pokročilých případech se rozvíjí mechanický syndrom s obrazem dušnosti, obtížného polykání, chrapotu při paréze laryngeálního nervu.
Při rozsáhlém postižení s nálezem vzdálených metastáz v játrech, plících či skeletu bývají popisovány typické těžké, imperativní průjmy.
Familiární varianty MTC se klinicky manifestují dříve než sporadické a postižení bývá typicky oboustranné. MTC v rámci syndromů MEN 2A či 2B se manifestuje obvykle jako první (pro vznik syndromu je totiž patognomický), nemusí to být ale pravidlem. V řadě případů se onemocnění primárně projeví feochromocytomem či hyperplazií příštítných tělísek, která je zejména u syndromu MEN 2A poměrně častá, ale klinicky může být málo výrazná nebo i zcela němá. U nemocných s agresivnějším syndromem MEN 2B je patrný typický fenotyp (marfanoidní habitus, silné malinovité rty, neurofi bromy na jazyku, případně na okraji víček, viscerální anomálie ve smyslu megacolon transversum a intestinální ganglioneuromatózy s poruchou pasáže, střídání průjmů se zácpou), takže diagnóza nebývá obtížná.
Diagnostika
Základní zobrazovací metodou je ultrasonografi cké vyšetření krku, doplněné případně punkcí tenkou jehlou s cytologickým vyšetřením, včetně imunocytochemickým (na přítomnost kalcitoninu, tyreoglobulinu a chromograninu A, který je u MTC pozitivní prakticky ve všech případech). Ostatní konvenční zobrazovací metody mají význam u pokročilých případů onemocnění se vzdálenými metastázami (počítačová tomografi e hrudníku a břicha, scintigrafi e skeletu, scintigrafi e99m-Tc-MIBI (methoxyisobutylisonitril), 123 I-MIBG(metajodobenzylguanidin), 111- In-Octreoscan (octreotid).
V případě podezření na mozkové metastázy využíváme vyšetření magnetickou rezonancí. U pacientů s vysokou hladinou kalcitoninu, při negativních předchozích vyšetřeních, je nezbytné doplnit o vyšetření pomocí 18F-FDG (fl uordeoxyglukózy) metodou hybridního zobrazení PET/CT (pozitronová emisní tomografi e).
MTC produkuje kalcitonin, jehož hladina v séru je citlivým nádorovým markerem. U nemocných s aktivním nádorem je jeho hodnota vždy značně vysoká (nad 1000 pg/ml), po zdařilé operaci klesá prakticky k nule. Kontrolní pooperační vyšetření kalcitoninémie provádíme vždy i se stimulačním testem v odstupu 3 měsíců po ope raci. Nález vyšších hladin kalcitoninu ukazuje na nedostatečnou radikalitu chirurgické léčby, případně na přítomnost metastatických ložisek, která je nutno hledat zobrazovacími metodami (ultrazvuk krku, CT hrudníku a břicha, scintigrafi cké metody).
Od poloviny 90. let minulého století se nedílnou součástí diagnostiky MTC staly metody molekulární genetiky. Familiární formy jsou charakterizovány zárodečnou bodovou mutací RET proto-onkogenu na 10. chromosomu. Význam genetického screeningu spočívá v tom, že stačí vyšetřit nemocného jedince s potvrzenou diagnózou MTC a až při pozitivním nálezu mutace jsou následně vyšetřenii jeho pokrevně příbuzní (napřed 1. linie, podle výsledků pak rozšíření testu na širší příbuzenstvo). Předtím bylo nutné odebírat krev na bazální a stimulovaný kalcitonin jak u nemocného, tak i u obou rodičů, sourozenců, dětí a tím stoupala jak náročnost na provedení a organizaci, tak i nákladnost vyšetření. U všech nemocných s MTC bylo nutno rovněž vyloučit či potvrdit přítomnost syndromu MEN 2, tedy feochromocytom a hyperparatyreózu. Vyšetření katecholaminů (v krvi i v moči), parathormonu, sérových hladin vápníku a anorganických fosfátů se v současné době rutinně provádí až při pozitivním genetickém vyšetření.
Nosiči mutací by měli být zařazeni do screeningového programu, při kterém je biochemické sledování hormonálně aktivních nádorů účinnou metodou časné diagnostiky. Efektivní léčba spočívá v andrenalektomii (u prokázaných případů MEN2B i jako preventivní výkon v časném věku) a totální tyreoidektomii.
Léčba
Základním léčebným opatřením u nemocných s MTC jak se sporadickou, tak
i u všech familiárních variant, je provedení radikálníhochirurgického výkonu. Je prokázáno, že nedostatečný, neradikální výkon zásadním způsobem ovlivňuje další prognózu onemocnění. Výkon má být proveden v rozsahu totální tyreoidektomie,
v případě pokročilého nálezu s postižením regionálních lymfatických uzlin na krku, rozšířený o modifi kovanou blokovou krční disekci. Není-li výkon dostatečný (dle klinického a zobrazovacího vyšetření, přetrvávání zvýšeného kalcitoninu) a při nemožnosti reoperace, je ve spolupráci s onkologem indikována zevní aktinoterapie na ložiska, případně (a to hlavně při generalizaci procesu) systémová chemoterapie. Je-li zjištěné metastatické ložisko dostupné chirurgické léčbě, je naší snahou provést operační výkon s následným onkologickým zajištěním (zevní aktinoterapie, chemoterapie). V poslední době se v oblasti léčby pokročilých MTC upírá pozornost
k metodám molekulární genetiky a genová terapie. Velmi optimistické jsou zprávy ohledně klinického zkoušení inhibitorů tyrozinkinázy a receptoru vaskulárního endoteliálního růstového faktoru(Vandetanib a další).
i u všech familiárních variant, je provedení radikálníhochirurgického výkonu. Je prokázáno, že nedostatečný, neradikální výkon zásadním způsobem ovlivňuje další prognózu onemocnění. Výkon má být proveden v rozsahu totální tyreoidektomie,
v případě pokročilého nálezu s postižením regionálních lymfatických uzlin na krku, rozšířený o modifi kovanou blokovou krční disekci. Není-li výkon dostatečný (dle klinického a zobrazovacího vyšetření, přetrvávání zvýšeného kalcitoninu) a při nemožnosti reoperace, je ve spolupráci s onkologem indikována zevní aktinoterapie na ložiska, případně (a to hlavně při generalizaci procesu) systémová chemoterapie. Je-li zjištěné metastatické ložisko dostupné chirurgické léčbě, je naší snahou provést operační výkon s následným onkologickým zajištěním (zevní aktinoterapie, chemoterapie). V poslední době se v oblasti léčby pokročilých MTC upírá pozornost
k metodám molekulární genetiky a genová terapie. Velmi optimistické jsou zprávy ohledně klinického zkoušení inhibitorů tyrozinkinázy a receptoru vaskulárního endoteliálního růstového faktoru(Vandetanib a další).
Asi u 1/3 nemocných se vzdálenými metastázami, zejména s metastatickým postižením jater, které je pro MTC typické a poměrně časté, má dobrou léčebnou odezvu nitrožilní infúze I-MIBG (metajodobenzylguanidin). Jde o syntetický analog norepinefrinu a guanetidinu. Při nálezu pozitivní akumulace v nádorovém ložisku (v rámci diagnostiky se používá 123I-MIBG) podáváme léčebnou dávku 131I-MIBG.
Léčba se opakuje v intervalu 6 až 12 měsíců. Dosavadní výsledky jsou poměrně uspokojivé, i když mají spíše paliativní charakter. Při neztišitelných průjmech a nezadržitelné progresi tumoru, přes výše uvedenou léčbu, lze v některých případech použít s dobrým terapeutickým efektem somatostatin a interferon. Léčba však musí být dlouhodobá až trvalá, je poměrně nákladná a s vedlejšími účinky (při dlouhodobé somatostatinové léčbě bývá hyperglykemie, steatorea, cholecystolitiáza, při aplikaci interferonu leukopenie a trombocytopenie s pseudochřipkovými projevy). Nemocní po operaci štítné žlázy dostávají tyreoidální substituci levothyroxinem, mělo by
být dosaženo eutyreózy, supresní léčba se nepoužívá (nezbytné je měření TSH, FT4).
Prognóza
Prognóza nemocných s medulárním karcinomem je horší než u pacientů s diferencovaným karcinomem, vycházejícím z folikulárních buněk, a je závislá na stadiu onemocnění v době diagnózy. Prognosticky nevýhodným faktorem je např. vyšší věk v době diagnózy (nad 60 let). Desetileté přežití se pohybuje kolem 75 %.
Významnou roli v prognóze nemoci má výsledek genetického vyšetření a typ nalezené mutace (např. nález mutace na kodonu 918 v 16. exonu je pokládán za prognosticky závažný, neboť je spojen s výskytem feochromocytomu v rámci syndromu MEN 2B). Lepší prognózu mají pacienti s familiární formou MEN 2A, pravděpodobně díky včasnější diagnóze těchto nemocných a pacienti diagnos-
tikovaní na základě aktivního screeningu.
Doc. MUDr. Petr Vlček, CSc., MHA
Klinika nukleární medicíny a endokrinologie 2. LF UK a FN Motol, Praha
originál článku se nachází na webu http://neuroendokrinni-nadory.cz/downloads/Somatuline_bulletin_3_2012.pdf


